Alte Pest-Genome geben Einblick in die genetische Geschichte der zweiten Pandemie

Ein internationales Forschungsteam hat kürzlich menschliche Überreste aus zehn archäologischen Stätten in England, Frankreich, Deutschland, Russland und der Schweiz analysiert. Ihr Ziel war es, Einblicke in die verschiedenen Stadien der zweiten Pestepidemie (14. bis 18. Jahrhundert) und die genetische Vielfalt des Pestbakteriums während und nach der Pandemie des „Schwarzen Todes“ zu gewinnen.
In einer in Nature Communications veröffentlichten Studie rekonstruierte das Team 34 Yersinia-pestis-Genome, um die genetische Geschichte des Bakteriums nachzuverfolgen. Dabei konnten wichtige Erkenntnisse über den Beginn und den Verlauf der zweiten Pestpandemie gewonnen werden.
Die zweite Pestpandemie begann Mitte des 14. Jahrhunderts mit dem „Schwarzen Tod“ und dauerte bis in das 18. Jahrhundert an. Den verheerenden Ausbrüchen in ganz Europa und in angrenzenden Regionen fielen schätzungsweise bis zu 60 Prozent der Bevölkerung zum Opfer. Aber woher kam dieser Stamm des Pesterregers Yersinia pestis? Und wie hat sich das Bakterium, nachdem es Europa erreicht hatte, weiterentwickelt und ausgebreitet?
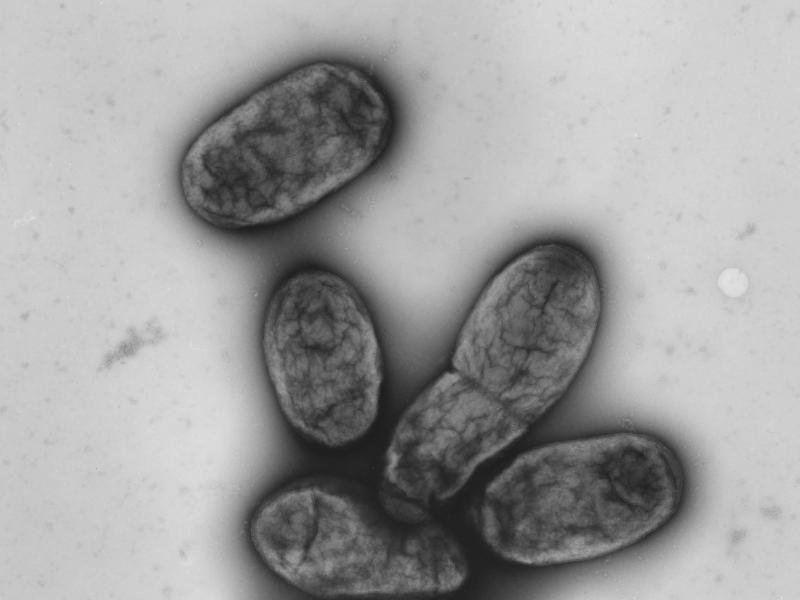
Diese elektronenmikroskopische Aufnahme zeigt das Pestbakterium Yersinia pestis. Foto: Robert-Koch-Institut/dpa/dpa
Y.-pestis-Stämme der zweiten Pandemie gehen auf einen einzigen Urahnen zurück
Bislang existieren nur wenige Daten zu frühen Ausbrüchen der Pest sowie ein Mangel an veröffentlichten genetischen Daten historischer Pest-Genome. Trotz der Allgegenwart des Schwarzen Todes in historischen Texten und im allgemeinen Bewusstsein sind einige Aspekte noch immer unklar. Von wo und über welche Route gelangte das Y. pestis-Bakterium zu dieser Zeit nach Europa? Wie verbreitete sich es sich über den Kontinent?
In der aktuellen Studie rekonstruierten die Wissenschaftler das Pest-Genom aus den Zähnen von 34 Pestopfern. Unter ihnen befinden sich auch zwei Individuen aus Laishevo, in der Wolga-Region Russlands. Bei der Analyse dieser Pest-Genome gelang es den Forschern, einen Bakterienstamm zu identifizieren, von dem alle weiteren Bakterienstämme der zweiten Pandemie abstammen. Darüber hinaus beobachtete das Team bei den Proben das Fehlen genetischer Vielfalt.
„Diese Ergebnisse deuten darauf hin, dass Yersinia pestis von Osten her nach Europa gelangte“, erklärt Ko-Autorin Maria Spyrou vom Jenaer Max-Planck-Institut für Menschheitsgeschichte (MPI-SHH). „Es ist jedoch möglich“, fährt sie fort, „dass zukünftig weitere Interpretationen möglich werden, wenn zum Beispiel eine bislang unbekannte genetische Vielfalt des Bakteriums in Westeurasien entdeckt würde.“

Massengrab aus der Zeit des Schwarzen Todes, an der archäologischen Stätte “16 rue des Trente Six Ponts” in Toulouse, Frankreich. Foto: Archeodunum SAS, Gourvennec Michaël
Fortbestand und Entwicklung des Y.-pestis-Bakteriums in Europa
Obwohl die Studie zeigt, dass der Schwarze Tod wahrscheinlich durch einen einzigen Stamm ausgelöst wurde, belegt die Analyse von Genomen aus der späteren Pandemie die Entstehung einer Linie mit einer höheren genetischen Vielfalt.
„In der späteren Phase der zweiten Pandemie sehen wir die Entwicklung zweier Zweige in Europa, was darauf hindeutet, dass das Pestbakterium in verschiedenen lokalen Reservoiren überlebte“, sagt Ko-Autor Marcel Keller vom MPI. „Moderne Nachfahren dieser Linie wurden jedoch bislang nicht gefunden, was auf ein Aussterben dieser Reservoire hindeutet.“
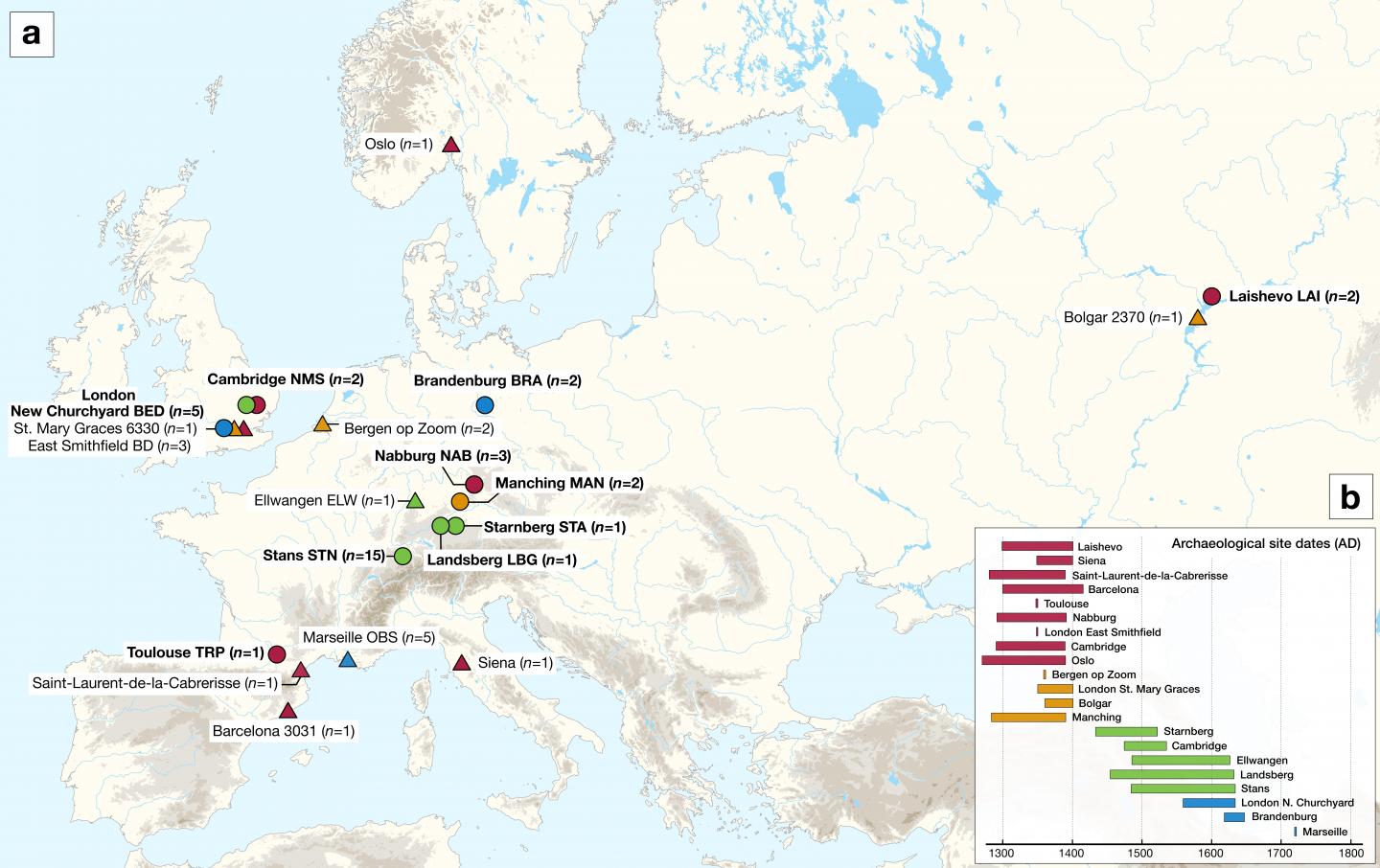
Die Karte zeigt die archäologischen Stätten der neu sequenzierten (Kreise) und bereits publizierter Pestgenome (Dreiecke), in zeitlicher Staffelung gefärbt. Foto: SPYROU ET AL./MPI-SHH
Das Team entdeckte außerdem in Genomen eines Zweigs den Verlust eines Erbgutteils. Darunter seien zwei Gene, die mit der Virulenz des Bakteriums in Verbindung stehen, also wie gefährlich bzw. ansteckend der Erreger ist. Interessanterweise wurde in Genomen der späten ersten Pandemie eine Deletion der gleichen Region festgestellt.
„Angesichts der Tatsache, dass diese Deletion in Linien der ersten und zweiten Pandemie auftritt, die beide inzwischen ausgestorben sind, wäre es wichtig in zukünftigen Studien zu klären, wie sich diese Gene auf das Überleben des Bakteriums in Menschen und Flöhen auswirken“, sagt Kirsten Bos, Forschungsgruppenleiterin am MPI.
Einblicke in die Mikroevolution eines Erregers
Die aktuelle Studie eröffnet neue Perspektiven auf den Beginn und Verlauf der zweiten Pandemie. Weiterhin ergänzt sie die Datenbank der veröffentlichten historischen Pest-Genome signifikant.

Maria Spyrou im Reinraumlabor des Jenaer Max-Planck-Instituts für Menschheitsgeschichte. Foto: Lyazzat Musralina
„Wir konnten zeigen, dass eine umfassende Analyse alter Y.-pestis-Genome einzigartige Einblicke in die Mikroevolution eines Erregers über einen Zeitraum von mehreren hundert Jahren liefern kann“, sagt Johannes Krause, Direktor der Abteilung für Archäogenetik am MPI und Leiter der Studie.
„In Zukunft wird die Integration dieser Daten in die Modellierung von Krankheitsausbrüchen, zusammen mit Daten aus anderen Disziplinen wie der Epidemiologie und Geschichte, für ein besseres Verständnis der zweiten Pandemie von Bedeutung sein.“ (MPI/ts)
Die PDF zur Studie finden Sie hier (Englisch): Phylogeography of the second plague pandemic revealed through analysis of historical Yersinia pestis genomes
















Prinzessin von Luxemburg: Shen Yun gibt „Hoffnung auf eine bessere Welt“

Krieg statt Klima: EU verschiebt ihre Prioritäten

Strahlendes Recycling: Unternehmen will Atommüll zur Energiequelle machen

Wende in der Außenpolitik durch Javier Milei: Argentinien will in die NATO


Kriminalstatistik: Verharmlosungstendenzen und eine Gleichung, die nicht aufgeht

„Der Alptraum geht wieder von vorn los“: Vater einer Amerikanerin von der chinesischen Polizei entführt

Ernüchternde Bilanz von Scholz in China


vielen Dank, dass Sie unseren Kommentar-Bereich nutzen.
Bitte verzichten Sie auf Unterstellungen, Schimpfworte, aggressive Formulierungen und Werbe-Links. Solche Kommentare werden wir nicht veröffentlichen. Dies umfasst ebenso abschweifende Kommentare, die keinen konkreten Bezug zum jeweiligen Artikel haben. Viele Kommentare waren bisher schon anregend und auf die Themen bezogen. Wir bitten Sie um eine Qualität, die den Artikeln entspricht, so haben wir alle etwas davon.
Da wir die Verantwortung für jeden veröffentlichten Kommentar tragen, geben wir Kommentare erst nach einer Prüfung frei. Je nach Aufkommen kann es deswegen zu zeitlichen Verzögerungen kommen.
Ihre Epoch Times - Redaktion